 Cholecystokinin is a peptide hormone of the gastrointestinal system responsible for stimulating the digestion of fat and protein. Cholecystokinin, previously called pancreozymin, is synthesized by I-cells in the mucosal epithelium of the small intestine and secreted in the duodenum, the first segment of the small intestine, and causes the release of digestive enzymes and bile from the pancreas and gallbladder, respectively. It also acts as a hunger suppressant. Recent evidence has suggested that it also plays a major role in inducing drug tolerance to opioids like morphine and heroin, and is partly implicated in experiences of pain hypersensitivity during opioid withdrawal.
Cholecystokinin is a peptide hormone of the gastrointestinal system responsible for stimulating the digestion of fat and protein. Cholecystokinin, previously called pancreozymin, is synthesized by I-cells in the mucosal epithelium of the small intestine and secreted in the duodenum, the first segment of the small intestine, and causes the release of digestive enzymes and bile from the pancreas and gallbladder, respectively. It also acts as a hunger suppressant. Recent evidence has suggested that it also plays a major role in inducing drug tolerance to opioids like morphine and heroin, and is partly implicated in experiences of pain hypersensitivity during opioid withdrawal.
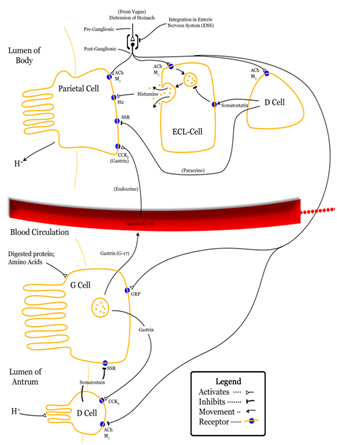
CCK mediates a number of physiological processes, including digestion and satiety. It is released by I cells located in the mucosal epithelium of the small intestine (mostly in the duodenum and jejunum), neurons of the enteric nervous system, and neurons in the brain. Release of CCK is stimulated by monitor peptide released by pancreatic acinar cells as well as CCK-releasing protein, a paracrine factor secreted by enterocytes in the gastrointestinal mucosa. In addition, release of acetylcholine by the parasympathetic nerve fibers of the vagus nerve also stimulates its secretion. The presence of fatty acids and/or certain amino acids in the chyme entering the duodenum is the greatest stimulator of CCK release.
CCK mediates digestion in the small intestine by inhibiting gastric emptying and gastric acid secretion. It stimulates the acinar cells of the pancreas to release digestive enzymes and stimulates the secretion of a juice rich in pancreatic digestive enzymes, hence the old name pancreozymin. Together these enzymes catalyze the digestion of fat, protein, and carbohydrates. Thus, as the levels of the substances that stimulated the release of CCK drop, the concentration of the hormone drops as well. The release of CCK is also inhibited by somatostatin and pancreatic peptide. Trypsin, a protease released by pancreatic acinar cells, hydrolyzes CCK-releasing peptide and monitor peptide, in effect turning off the additional signals to secrete CCK.
As a peptide hormone, CCK mediates satiety by acting on the CCK receptors distributed widely throughout the central nervous system. In humans, it has been suggested that CCK administration causes nausea and anxiety, and induces a satiating effect. CCK-4 is routinely used to induce anxiety in humans though certainly different forms of CCK are being shown to have highly variable effects.[5] The mechanism for this hunger suppression is thought to be a decrease in the rate of gastric emptying.[6]
CCK also has stimulatory effects on the vagus nerve, effects that can be inhibited by capsaicin.[7] The stimulatory effects of CCK oppose those of ghrelin, which has been shown to inhibit the vagus nerve.[8] The CCK tetrapeptide fragment CCK-4 (Trp-Met-Asp-Phe-NH2) reliably causes anxiety when administered to humans, and is commonly used in scientific research to induce panic attacks for the purpose of testing new anxiolytic drugs.
In recent years, the researchers pay more and more attention to the the relationship between Cholecystokinin and the obesity. For example, Diet-induced obesity accelerates the appearance of CCK-resistance as well as the return of high sensitivity to CCK in further aging, while chronic calorie-restriction prevents the development of resistance, as if the speed of the age-related regulatory changes was altered by the nutritional state. The effects of ICV applied CCK also change with age: the characteristic anorexic and hypermetabolic/hyperthermic effects can be observed in young adult rats, but the effects gradually and monotonically decline with age and disappear by the old age of 24 months. These disparate age-related patterns of CCK efficacy upon peripheral or central administration routes may indicate that although both peripheral and central CCKR-s exert anorexic effects, they may have dissimilar roles in the regulation of overall energy balance.
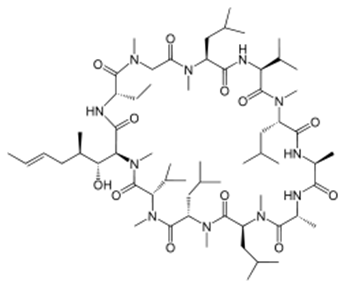
Karebay can synthetic Cholecystokinin with sulfoacid and fluorescent labels, so that scientists can knew more details about its molecular mechanism.
Karebay (www.karebaybio.com) has a professional team devoted to biotechnological products and development. We offer high-quality peptide synthesis products for sale around the world, including over 1,000 catalog peptides, and nearly 100 pharmaceutical peptides and cosmetic peptides products.